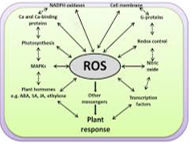
Hydrogen peroxide is noteworthy because it readily permeates membranes and it is therefore not compartmentalised in the cell. Numerous enzymes (peroxidases) use hydrogen peroxide as a substrate in oxidation reactions involving the synthesis of complex organic molecules. The well-known reactivity of hydrogen peroxide is not due to its reactivity per se, but requires the presence of a metal reductant to form the highly reactive hydroxyl radical which is the strongest oxidizing agent known and reacts with organic molecules at diffusion-limited rates.
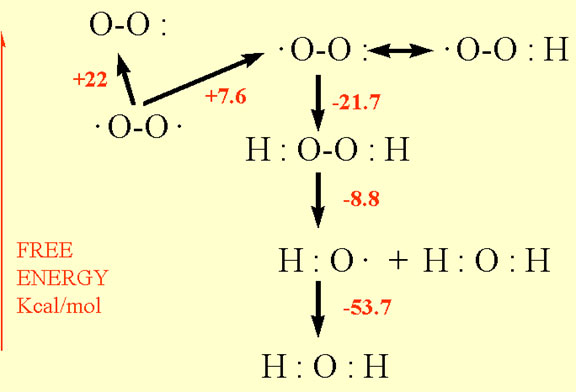
Fenton described in the late nineteenth century (Fenton, 1894; 1899) the oxidising potential of hydrogen peroxide mixed with ferrous salts. Forty years later, Haber and Weiss (1934) identified the hydroxyl radical as the oxidising species in these reactions:
(1) ![]()
In biological systems the availability of ferrous ions limits the rate of reaction, but the recycling of iron from the ferric to the ferrous form by a reducing agent can maintain an ongoing Fenton reaction leading to the generation of hydroxyl radicals. One suitable reducing agent is superoxide which participates in the overall reaction 2 as two half reactions shown in reactions 3 and 4:
(2) ![]()
(3) ![]()
(4) ![]()
Therefore, in the presence of trace amounts of iron, the reaction of superoxide and hydrogen peroxide will form the destructive hydroxyl radical and initiate the oxidation of organic substrates. Metals other than iron may also participate in these electron transfer reactions by cycling between oxidised and reduced states.
The oxidation of organic substances may proceed by two possible reactions Ä addition of OH to the organic molecule, or abstraction of a hydrogen atom from it. In the addition reaction (reaction 5), the hydroxyl radical adds to an organic substrate forming a hydroxylated product that is further oxidised by ferrous ions, oxygen or other agents to a stable, oxidised product (reactions 6 and 7). The hydroxylated products can also dismutate to form cross-linked products (reaction 8).
(5) ![]()
(6) ![]()
(7) ![]()
(8) ![]()
In the abstraction reaction, the hydroxyl radical oxidises the organic substrate forming water and an organic radical (reaction 9). The latter product has a single unpaired electron and thus can react with oxygen in the triplet ground-state (reaction 10). The addition of triplet oxygen to the carbon radical can lead to the formation of a peroxyl radical which can readily abstract hydrogen from another organic molecule leading to the formation of a second carbon radical (reaction 11). This chain reaction is why oxygen free radicals cause damage far in excess of their initial concentration.
(9) ![]()
(10) ![]()
(11) ![]()
BIOLOGICAL REACTIONS OF OXYGEN RADICALS
The reactions of activated oxygen with organic substrates are complex even in vitro with homogenous solutions, but in biological systems there are even more complications due to the surface properties of membranes, electrical charges, binding properties of macromolecules, and compartmentalisation of enzymes, substrates and catalysts. Thus, various sites even within a single cell differ in the nature and extent of reactions with oxygen.
The nature of the oxidative injury that causes cell death is not always obvious. The mechanisms by which oxygen radicals damage membrane lipids are well accepted, and consequently oxidative damage is often exclusively associated with these peroxidation reactions in membrane lipids. What is sometimes overlooked in our research on environmental stress in plants is that activated forms of oxygen also degrade proteins and nucleic acids, reactions which can also be very lethal. In this section some of the major reactions of activated oxygen with lipids, protein, and nucleic acids are reviewed.
OXIDATIVE DAMAGE TO LIPIDS
Classical Peroxidation Reactions
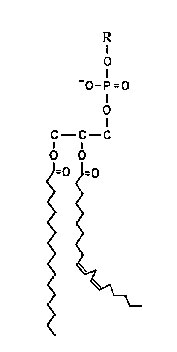
The reactions of oxygen free radicals with polyunsaturated lipids have been extensively researched because of their involvement in rancidity and the development of undesirable odours and flavours in foods. Historically these reactions are the most frequently cited consequence of oxygen radical production in plant cells. Perhaps the mechanisms were so well established by oil chemists long before the recognition of their importance in biology that plant biologists applied these mechanisms directly to their experimental systems, rarely questioning their validity or transposability. This has delayed recognition of the presence of free radical reactions in plant membranes. The complexity of the biological membrane is well established and the reader is referred elsewhere for more detailed considerations of its structure (Leshem, 1992). The lipid bilayer membrane is composed of a mixture of phospholipids and glycolipids that have fatty acid chains attached to carbon 1 and 2 of the glycerol backbone by an ester linkage. The peroxidation reactions differ among these fatty acids depending on the number and position of the double bonds on the acyl chain and the reader is referred to Frankel (1985) for a detailed review. The following is a simplified summary of these reactions for a general lipid, `R’, and for a specific fatty acid, linoleate, which is common in plant cell membranes.
The peroxidation of lipids involves three distinct steps: initiation, propagation and termination. The initiation reaction between an unsaturated fatty acid (e.g. linoleate) and the hydroxyl radical involves the abstraction of an H atom from the methylvinyl group on the fatty acid (reaction 9); in the case of linoleate this occurs at carbon-11 (Fig. 3). The remaining carbon centred radical, forms a resonance structure sharing this unpaired electron among carbons 9 to 13. In the propagation reactions, this resonance structure reacts with triplet oxygen, which remember is a biradical having two unpaired electrons and therefore reacts readily with other radicals. This reaction forms a peroxy radical (reaction 10). In the case of linoleate, addition occurs at either carbon-9 or -13 (Fig 3). The peroxy radical then abstracts an H atom from a second fatty acid forming a lipid hydroperoxide and leaving another carbon centred free radical (reaction 11) that can participate in a second H abstraction (reaction 10). Therefore, once one hydroxyl radical initiates the peroxidation reaction by abstracting a single H atom, it creates a carbon radical product (R) that is capable of reacting with ground state oxygen in a chain reaction. The role of the hydroxyl radical is analogous to a “spark” that starts a fire. The basis for the hydroxyl radical’s extreme reactivity in lipid systems is that at very low concentrations it initiates a chain reaction involving triplet oxygen, the most abundant form of oxygen in the cell.
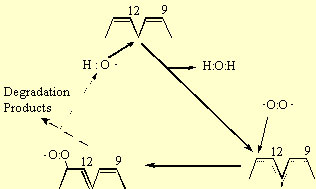
The lipid hydroperoxide (ROOH) is unstable in the presence of Fe or other metal catalysts because ROOH will participate in a Fenton reaction leading to the formation of reactive alkoxy radicals:
(12) ![]()
Therefore, in the presence of Fe, the chain reactions are not only propagated but amplified. Note that two radicals are produced by the summation of reactions 9 to 12. Among the degradation products of ROOH are aldehydes, such as malondialdehyde, and hydrocarbons, such as ethane and ethylene, that are commonly measured end products of lipid peroxidation.
The peroxidation reactions in membrane lipids are terminated when the carbon or peroxy radicals cross-link to form conjugated products that are not radicals, such as those shown in reactions 13 to 15:
(13) ![]()
(14) ![]()
(15) ![]()
Typically, high molecular weight, cross-linked fatty acids and phospholipids accumulate in peroxidised membrane lipid samples.
Singlet oxygen can react readily with unsaturated fatty acids producing a complex mixture of hydroperoxides. Again, the chemistry of these reactions is based on foods (Bradley and Minn, 1992). Oxidation of unsaturated fatty acids by singlet oxygen produces distinctly different products than the hydroxyl radical (Bradley and Minn, 1992). Once formed the lipid hydroperoxides will decompose into a variety of products, some of which can produce oxygen free radicals in the presence of metal catalysts (reaction 12).
Unique Reactions in Plant Membranes
The above mechanisms predict that oxygen free radical or lipid peroxidation reactions in plant membranes would selectively degrade unsaturated fatty acids and accumulate aldehydes, hydrocarbons, and cross-linked products. When examining the effects of environmental stresses on plant membranes, many studies have measured the products of lipid peroxidation, such as malondialdehyde and/or ethane and concluded that oxygen free radicals are involved in these stress responses. When the substrates of these reactions, the membrane fatty acids, have been examined, it has been very often observed that the unsaturated fatty acids are not selectively degraded, and therefore these reports have concluded that oxygen free radicals are not involved in these stress responses. This controversy has caused many to rule out the involvement of oxygen free radicals in processes such as seed ageing (Wilson and McDonald, 1986). However, in vitro experiments that have treated plant membranes with Fenton reaction products have shown that degradation of plant membrane lipids by oxygen free radicals does not involve selective loss of unsaturated fatty acids. For example, in Table 1, microsomal membranes isolated from wheat (Triticum aestivum) crowns and liposomes prepared from a commercial preparation of soybean asolecithin were treated in vitro with oxygen radicals generated by Fe-ascorbate. In both samples, there was destruction of fatty acids and their recovery from solution was lower after the free radical treatment. In the liposome sample there was selective degradation of unsaturated fatty acids; the proportion of linoleic and linolenic acids relative to the other fatty acids declined. In contrast, treatment of the wheat microsomal membranes caused degradation og the phospholipids but no change in the proportion of the fatty acids. Inother words, there was not selective degradation of the unstaurated fatty acids. Clearly other reactions to those described above were occurring in these plant membranes.
An alternative to the classical mechanism of lipid peroxidation was proposed by Niehaus (1978) based on his observation that most esters react with superoxide by cleaving the C-O bond. Since the fatty acid chains are attached to the glycerol backbone of the phospholipid molecule by an ester bond (Fig. 4), superoxide attack on a phospholipid bilayer would produce free fatty acids by de-esterification reactions. This was experimentally observed by Senaratna et al. (1985) in microsomal membranes from soybean seed axes treated in vitro with superoxide from xanthine oxidase.
Table 1: Degradation of phospholipid and esterified fatty acid in two membrane systems by Fenton reaction products. Data for each fatty acid are expressed as its proportion (%) of all fatty acids recovered (i.e.each sample totals 100%). Phospholipid (PL) recovery is expressed as a % of the original PL before treatment. Adapted from McKersie et al. (1990) | ||||
Fatty Acid | Liposomes | Wheat Microsomes | ||
Before | After | Before | After | |
16:0 | 21 | 34 | 29 | 30 |
18:0 | 3 | 5 | 1 | 1 |
18:1 | 7 | 10 | 6 | 7 |
18:2 | 61 | 47 | 28 | 29 |
18:3 | 8 | 4 | 37 | 35 |
PL Recovery | 100 | 51 | 100 | 65 |
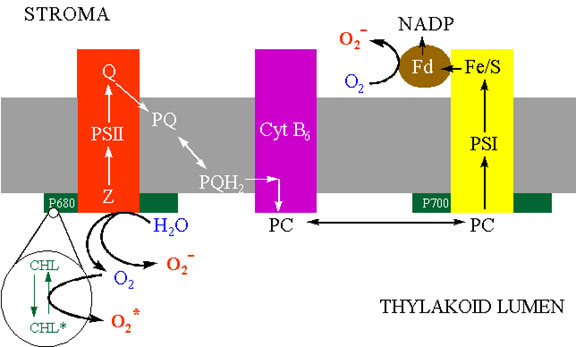
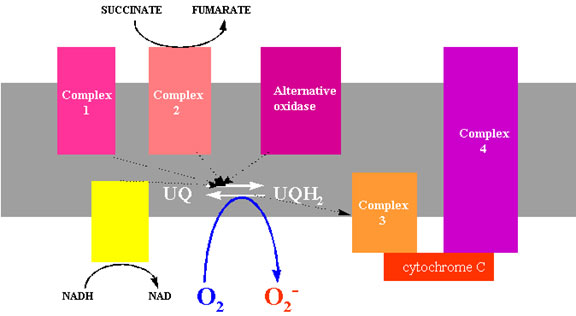
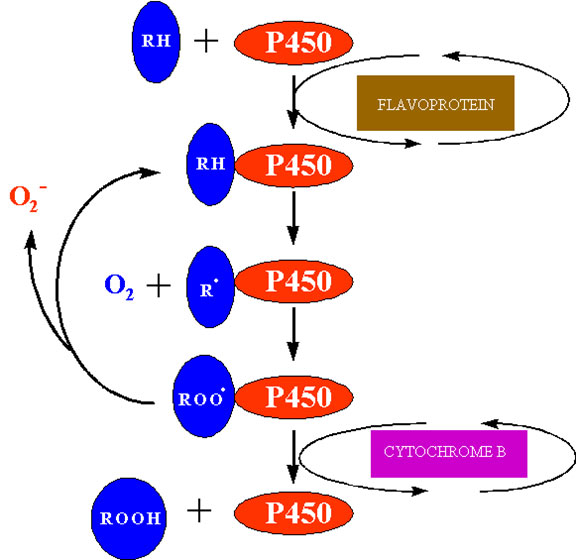
ENDOPLASMIC RETICULUM
Various oxidative processes, including oxidation, hydroxylations, dealkylations, deaminations, dehalogenation and desaturation, occur on the smooth endoplasmic reticulum. Mixed function oxygenases that contain a heme moiety add an oxygen atom into an organic substrate using NAD(P)H as the electron donor. The generalised reaction catalysed by cytochrome P450 is:
(17) ![]()
The best characterised cytochrome P450 in plants is cinnamate-4-hydroxylase which functions in flavonoid and lignin biosynthesis, but other mixed function oxidases function in other biochemical pathways including gibberellin and sterol biosynthesis. Activation of oxygen by these systems is an essential prerequisite to oxygen addition reactions in the synthesis of these “complex” metabolites. Superoxide is produced by microsomal NAD(P)H dependent electron transport involving cytochrome P450 (Winston and Cederbaum, 1983). One possible site at which this may occur is shown in figure 7. After the univalent reduction of the substrate (RH) and the addition of triplet oxygen to form the complex P450 – RHOO the complex may decompose to P450-RH and release superoxide.
MICROBODIES
Peroxisomes and glyoxysomes are organelles with a single membrane that compartmentalises enzymes involved in the ß-oxidation of fatty acids, and the glyoxylic acid cycle including glycolate oxidase, catalase and various peroxidases. Glycolate oxidase produces H2O2 in a two electron transfer from glycolate to oxygen (Lindqvist et al., 1991). Xanthine oxidase, urate oxidase and NADH oxidase generate superoxide as a consequence of the oxidation of their substrates. The xanthine oxidase reaction is often used in vitro as a source of superoxide producing one mole of superoxide during the conversion of xanthine to uric acid (Fridovich, 1970).
PLASMA MEMBRANES
A superoxide-generating NAD(P)H oxidase activity has been clearly identified in plasmalemma enriched fractions (Vianello and Macri, 1991). These flavoproteins may produce superoxide by the redox cycling of certain quinones or nitrogenous compounds. In the root, NAD(P)H oxidase reduces Fe3+ to Fe2+ converting it to a form that can be transported. Dysfunction of this root enzyme will produce superoxide (Cakmak and Marschner, 1988). An auxin-activated NADH oxidase has been associated with acidification of the cell wall and auxin-stimulated cell elongation (Morré et al., 1988).
The plant NAD(P)H oxidase may have an analogous function to the animal enzyme. Leucocytes contain an NADH oxidase on the outer membrane surface which is activated in response to a foreign agent, generating superoxide that initiates oxidative reactions that destroy the potential pathogen (Hohn and Lehere, 1975). In plants, fungal elicitors cause a similar formation of superoxide that has been linked to the hypersensitive response to some pathogenic fungi (Doke and Ohashi, 1988; Doke et al., 1991). Wounding, heat shock and xenobiotics transiently activate this superoxide generating reaction, and consequently, it has been proposed that these superoxide generating reactions may serve as a signal in plant cells to elicit responses to biological, physical or chemical stress (Doke et al., 1991).
CELL WALLS
Although it is not immediately obvious, cell walls are active sites of metabolism, and also oxygen activation. Some of these reactions may be involved in the defense reactions against pathogens as described above. Others may involve the degradation or compartmentation of xenobiotic chemicals. However, the most common reactions are biosynthetic. For example, the phenylpropanoid precursors of lignin are crosslinked by H2O2 dependent reactions, that randomly link the subunits to form lignin (Gross, 1980). NADH is generated by a cell wall malate dehydrogenase, and then used to form H2O2 (Gross et al., 1977), possibly by the NADH oxidase on the plasmalemma (Vianello and Macri, 1991). Diamine oxidases are also involved in production of activated oxygen in the cell wall using diamines or polyamines (putrescine, spermidine, cadaverine, etc.) to reduce a quinone that will autoxidize, forming peroxides (Vianello and Macri, 1991; Elstner, 1991).
DEFENCE MECHANISMS
SUPEROXIDE DISMUTASE
Superoxide dismutase (SOD) was first isolated by Mann and Keilis (1938) and thought to be a copper storage protein. Subsequently, the enzyme was identified by a number of names, erythrocuprein, indophenol oxidase, and tetrazolium oxidase until its catalytic function was discovered by McCord and Fridovitch (1969). SOD is now known to catalyse the dismutation of superoxide to hydrogen peroxide and oxygen:
(18) 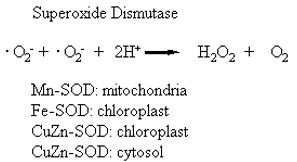
Therefore, the activity of this enzyme determines the relative proportions of the two constituents of the Haber-Weiss reaction that generates hydroxyl radicals (reaction 2). Since SOD is present in all aerobic organisms and most (if not all) subcellular compartments that generate activated oxygen, it has been assumed that SOD has a central role in the defence against oxidative stress (Beyer, et al., 1991; Bowler et al., 1992; Scandalias, 1993). There are three distinct types of SOD classified on the basis of the metal cofactor: the copper/zinc (Cu/Zn – SOD), the manganese (Mn-SOD) and the iron (Fe-SOD) isozymes (Bannister et al., 1987). These isozymes can be separated by native polyacrylamide gel electrophoresis, their activity detected by negative staining and identified on the basis of their sensitivity to KCN and H2O2. The Mn-SOD is resistant to both inhibitors, whereas the Cu/Zn-SOD is sensitive to both inhibitors; Fe-SOD is resistant to KCN, and sensitive to H2O2. The subcellular distribution of these isozymes is also distinctive. The Mn-SOD is found in the mitochondria of eukaryotic cells; some Cu/Zn-SOD isozymes are found in the cytosol, others in the chloroplasts of higher plants. The Fe-SOD isozymes are often not detected in plants, but when detected, Fe-SOD is usually associated with the chloroplast compartment (Bowler et al., 1992). The prokaryotic Mn-SOD and Fe-SOD, and the eukaryotic Cu/Zn-SOD enzymes are dimers, whereas the Mn-SOD of mitochondria are tetramers (Scandalias, 1993). Peroxisomes and glyoxysomes of watermelons (Citrillus vulgaris) have been shown to contain both Cu/Zn- and Mn-SOD activity (Sandalio and Del Rio, 1988), but there are no reports of extracellular SOD enzymes in plants. All forms of the SOD are nuclear-encoded and are targeted to their respective subcellular compartments by an amino terminal targeting sequence. Several forms of SOD have been cloned from a variety of plants (Scandalias, 1990; Bowler, 1992).
Prokaryotic cells, and many eukaryotic algae contain only the Mn-SOD and Fe-SOD isozymes which are believed to be more ancient forms. In the bacteria E. coli, SOD activity is transcriptionally regulated by the SOX RS operon (Farr and Kogoma, 1991) but investigations into the regulatory mechanism of SOD expression in plants are only beginning (Bowler et al., 1992). To date it has been shown that SOD activity is increased in cells in response to diverse environmental and xenobiotic stresses including paraquat, high light, waterlogging and drought. Apparently, each of the SOD isozymes are independently regulated according to the degree of oxidative stress experienced in the respective subcellular compartments, but how this is communicated at the molecular level is unknown. Bowler et al (1992) have suggested that this role may be served by unique lipid peroxidation products from each organelle that diffuse from the site of oxidative damage to the nucleus where they would enhance transcription of specific SOD genes.
Several reviews on superoxide dismutase have recently been published which describe the characteristics of the enzymes, the cloned cDNA sequences and genes, and the effects of overexpression in transgenic plants (Bowler, et al., 1994, Doke, et al., 1994, Foyer, et al., 1994, Gressel and Galun, 1994, Scandalios, 1993, Van Camp, et al., 1994)
CATALASE
Catalase is a heme-containing enzyme that catalyses the dismutation of hydrogen peroxide into water and oxygen. The enzyme is found in all aerobic eukaryotes and is important in the removal of hydrogen peroxide generated in peroxisomes (microbodies) by oxidases involved in ß-oxidation of fatty acids, the glyoxylate cycle (photorespiration) and purine catabolism. Catalase was one of the first enzymes to be isolated in a highly purified state. All forms of the enzyme are tetramers in excess of 220,000 molecular weight. Multiple forms of catalase have been described in many plants. These forms have been cloned from maize (Redinbaugh et al., 1988; Scandalias, 1990) and homologous genes has been cloned from several other plants. Maize has three isoforms termed cat-1, cat-2 and cat-3, that are on separate chromosomes and are differentially expressed and independently regulated (Scandalias, 1990). Cat-1 and cat-2 are localised in peroxisomes and the cytosol, whereas cat-3 is mitochondrial. Careful examination of the structure of beef liver catalase has shown four NADPH binding sites per catalase tetramer (Fita and Rossmann, 1985), but these sites were not in close association with the hydrogen peroxide reaction centre. Instead, NADPH functions in animal catalase to protect against inactivation by hydrogen peroxide (Kirkman et al., 1987). The only plant catalase examined, potato, does not contain NADPH (Beaumont et al., 1990). It is interesting in this regard to note that catalase is very sensitive to light and has a rapid turnover rate similar to that of the D1 protein of PSII (Hertwig et al., 1992). This may be a result of light absorption by the heme group or perhaps hydrogen peroxide inactivation. Regardless, stress conditions which reduce the rate of protein turnover, such as salinity, heat shock or cold, cause the depletion of catalase activity (Hertwig et al., 1992; Feirabend et al., 1992). This may have significance in the plant’s ability to tolerate the oxidative components of these environmental stresses.
ASCORBIC ACID
L-ascorbic acid (vitamin C) is an important vitamin in the human diet and is abundant in plant tissues. Green leaves have the same amount of ascorbate as chlorophyll. Because of its nutritional importance, the distribution of ascorbate has been extensively quantified in plants; however, relatively little consideration has been given to its function in the plant. Ascorbate has been shown to have an essential role in several physiological processes in plants, including growth, differentiation and metabolism (Foyer, 1993). Ascorbate functions as a reductant for many free radicals, thereby minimising the damage caused by oxidative stress but ascorbate may have other functions which remain undefined.
Apparently synthesis of ascorbate occurs in the cytosol because a specific ascorbate translocator has been identified on the chloroplast envelope. L-ascorbic acid is synthesised from hexose sugars in higher plants but controversy remains concerning some steps in its synthesis (Loewus, 1988). Although two distinct pathways are possible (Foyer, 1993), higher plants primarily convert D-glucose to ascorbate by a direct conversion that maintains the carbon chain in the same sequence. The pathway involves the oxidation of carbon-1 of D-glucose and enediol formation between carbons 2 and 3:
(19) D-glucose D-glucosone L-sorbosone L-ascorbic acid
Ascorbate can directly scavenge oxygen free radicals with and without enzyme catalysts and can indirectly scavenge them by recycling tocopherol to the reduced form. By reacting with activated oxygen more readily than any other aqueous component, ascorbate protects critical macromolecules from oxidative damage. The reaction with the hydroxyl radical is limited only by diffusion.
The reaction with superoxide may serve a physiologically similar role to SOD:
(20)
2 O 2 + 2H+ + ascorbate 2H2O2 + dehydroascorbate
The reaction with hydrogen peroxide is catalysed by ascorbate peroxidase (Asada, 1992):
(21)
H2O2 + 2 ascorbate 2H2O + 2 monodehydroascorbate
The indirect role of ascorbate as an antioxidant is to regenerate membrane-bound antioxidants, such as a-tocopherol, that scavenge peroxyl radicals and singlet oxygen, respectively:
(22)
tocopheroxyl radical + ascorbate -tocopherol + monodehydroascorbate
The above reactions indicate that there are two different products of ascorbate oxidation, monodehydroascorbate and dehydroascorbate, that represent one and two electron transfers, respectively (Fig. 9). The monodehydroascorbate can either spontaneously dismutate (reaction 23) or is reduced to ascorbate by NAD(P)H monodehydroascorbate reductase (reaction 24):
(23) 2 monodehydroascorbate ascorbate + dehydroascorbate
(24) monodehydroascorbate + NAD(P)H ascorbate + NAD(P)
The dehydroascorbate is unstable at pH greater than 6 decomposing into tartrate and oxalate. To prevent this, dehydroascorbate is rapidly reduced to ascorbate by dehydroascorbate reductase using reducing equivalents from glutathione (GSH):
(25)
2 GSH + dehydroascorbate GSSG + ascorbate
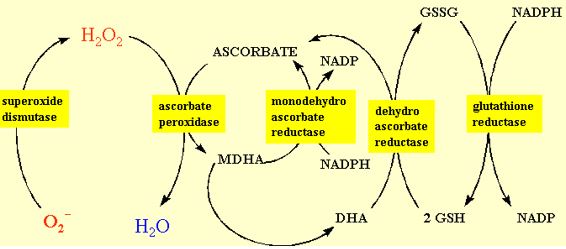
Ascorbate has been found in the chloroplast, cytosol, vacuole and extra-cellular compartments of the cell. About 20-40% of the ascorbate in the mesophyll leaf cell is in the chloroplast. The chloroplast contains all the enzymes to regenerate reduced ascorbate from its oxidised products. Foyer and Halliwell (1976) proposed that hydrogen peroxide was dissipated in the chloroplast by the coupling of ascorbate and glutathione redox cycling as shown in figure 10. Many of the details of this pathway and characterisation of the enzymes has been conducted by K. Asada in Japan. Consequently, this sequence of reactions is referred to as the Halliwell-Asada pathway. Illuminated chloroplasts produce superoxide and hydrogen peroxide from sites on the thylakoids, most commonly PSI. Superoxide is converted into hydrogen peroxide by either spontaneous dismutation or by the SOD enzyme. Hydrogen peroxide is scavenged by ascorbate and the enzyme ascorbate peroxidase (Asada, 1992). The monodehydroascorbate has two routes of regeneration, one via monodehydroascorbate reductase, the other via dehydroascorbate reductase and glutathione. The terminal electron donor is NADPH. This pathway serves two functions. One is the detoxification of hydrogen peroxide that might otherwise participate is Fenton reactions, and the second in the oxidation of NADPH. The latter function is an apparently energy-consuming, wasteful process analogous to photorespiration. It might at first seem more logical that the chloroplast contain catalase because it would allow the dissipation of hydrogen peroxide without “wasting” NADPH. However, it should be realised that conditions favouring electron transfer from PSI to oxygen generally cause a high redox potential, i.e. high NADPH/NADP ratio. By reducing this redox potential through the Halliwell-Asada pathway, the tendency of PSI to reduce oxygen is minimised.